
Position-specific isotope analysis
Position-specific isotope analysis, also called site-specific isotope analysis, is a branch of isotope analysis aimed at determining the isotopic composition of a particular atom position in a molecule. Isotopes are elemental variants with different numbers of neutrons in their nuclei, thereby having different atomic masses. Isotopes are found in varying natural abundances depending on the element; their abundances in specific compounds can vary from random distributions (i.e., stochastic distribution) due to environmental conditions that act on the mass variations differently. These differences in abundances are called "fractionations," which are characterized via stable isotope analysis.
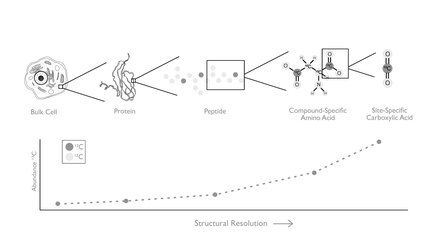
Isotope abundances can vary across an entire substrate (i.e., “bulk” isotope variation), specific compounds within a substrate (i.e., compound-specific isotope variation), or across positions within specific molecules (i.e., position specific isotope variation). Isotope abundances can be measured in a variety of ways (e.g., isotope ratio mass spectrometry, laser spectrometry, NMR, ESI-MS). Early analyses varied in technique, but were commonly limited by their ability to only measure average isotope compositions over molecules or samples. While this allows isotope analysis of the bulk substrate, it eliminates the ability to distinguish variation between different sites of the same element within the molecule. The field of position-specific isotope biogeochemistry studies these intramolecular variations, known as “position-specific isotope” and “site-specific isotope” enrichments. It focuses on position-specific isotope fractionations in many contexts, development of technologies to measure these fractionations and the application of position-specific isotope enrichments to questions surrounding biogeochemistry, microbiology, enzymology, medicinal chemistry, and earth history.
Position-specific isotope enrichments can retain critical information about synthesis and source of the atoms in the molecule. Indeed, bulk isotope analysis averages site-specific isotope effects across the molecule, and so while all those values have an influence on the bulk value, signatures of specific processes may be diluted or indistinguishable. While the theory of position-specific isotope analysis has existed for decades,[1] new technologies exist now to allow these methods to be much more common.[2] The potential applications of this approach are widespread, such as understanding metabolism in biomolecules, environmental pollutants in air, inorganic reaction mechanisms, etc. Clumped isotope analysis, a subset of position-specific isotope analysis, has already proven useful in characterizing sources of methane, paleoenvironment, paleoaltimetry, among many other applications. More specific case studies of position-specific isotope fractionation are detailed below.
Theory
[edit]Stable isotopes do not decay, and the heavy and light isotope masses affect how they partition within the environment. Any deviation from a random distribution of the light and heavy isotopes within the environment is called fractionation, and consistent fractionations as a result of a particular process or reaction are called "isotope effects."
Isotope Effects
[edit]Isotope effects are recurring patterns in the partitioning of heavy and light isotopes across different chemical species or compounds, or between atomic sites within a molecule. These isotope effects can come about from a near infinite number of processes, but most of them can be narrowed down into two main categories, based on the nature of the chemical reaction creating or destroying the compound of interest:
(1) Kinetic isotope effects manifest in irreversible reactions, when one isotopologue is preferred in the transition state due to the lowest energy state. The preferred isotopologue will depend on whether the transition state of the molecule during a chemical reaction is more like the reactant or the product. Normal isotope effects are defined as those which partition the lighter isotope into the products of the reaction. Inverse isotope effects are less common as they preferentially partition the heavier isotope into the products.
(2) Equilibrium isotope effects manifest in reversible reactions, when molecules can exchange freely to reach the lowest possible energy state.
These variations can occur on a compound-specific level, but also on a position-specific level within a molecule. For instance, the carboxyl site of amino acids is exchangeable and therefore its carbon isotope signature can change over time and may not represent the original carbon source of the molecule.
Biological fractionation
[edit]
Chemical reactions in biological processes are controlled by enzymes that catalyze the conversion of substrate to product. Since enzymes can alter the transition state structure for reactions, they also change kinetic and equilibrium isotope effects. Placed in the context of a metabolism, the expression of isotope effects on biomolecules is further controlled by branch points. Different pathways of biosynthesis will use different enzymes, yielding a range of position specific isotope enrichments. This variability allows position-specific isotope measurements to discern multiple biosynthetic pathways from the same metabolic product.[3] Biogeochemists use position specific isotope enrichments from amino acids, lipids, and sugars in nature to interpret the relative importance of different metabolisms.
Mechanism
[edit]The position-specific isotope effect of an enzymatic reaction is expressed as the ratio of rate constants for a monoisotopic substrate and a substrate substituted with one rare isotope. For example, enzyme formate dehydrogenase catalyzes the reaction of formate and NAD+ to carbon dioxide and NADH. The hydrogen of formate is directly transferred to NAD+. This step has an isotope effect, because the rate of protium transfer from formate to NAD+ is nearly three times faster than the rate of the same reaction with a deuterium transfer. This is also an example of a primary isotope effect.[4] A primary isotope effect is one in which the rare isotope is substituted where a bond is broken or formed. Secondary isotope effects occur on other positions in the molecule and are controlled by the molecular geometry of the transition state. These are generally considered to be negligible but do arise in certain cases, especially for hydrogen isotopes.[4]
Unlike abiotic reactions, enzymatic reactions occur through a series of steps, including substrate-enzyme binding, conversion of substrate to product, and dissociation of enzyme-product complex. The observed isotope effect of an enzyme will be controlled by the rate limiting step in this mechanism. If the step that converts substrate to product is rate limiting, the enzyme will express its intrinsic isotope effect, that of the bond forming or breaking reaction.[5]
Abiological fractionation
[edit]Like biotic molecules, position specific isotope enrichments in abiotic molecules can reflect the source of chemical precursors and synthesis pathways. The energy for abiotic reactions can come from many different sources, which will affect fractionation. For instance, metal catalysts can speed up abiotic reactions. Reactions can be slowed down or sped up by different temperature and pressure conditions, which will affect the equilibrium constant or activation energy of reversible and irreversible reactions, respectively.
For example, carbon in the interstellar medium and solar nebula partition into distinct states based on thermodynamic favorability. Measuring site-specific isotope enrichments of carbon from organic molecules extracted from carbonaceous chondrites can elucidate where each carbon atom comes from, and how organic molecules can be synthesized abiotically.[6] More broadly, these isotope enrichments can provide information about physical processes in the region where the molecular precursors were formed, and where the molecule formed in the solar system (i.e., nucleosynthetic heterogeneity, mass independent fractionation, self-shielding, etc.).
Another example of distinct site-specific fractionations in abiotic molecules is Fischer-Tropsch-type synthesis, which is thought to produce abiogenic hydrocarbon chains.[6] Through this reaction mechanism, site enrichments of carbon would deplete as carbon chain length increases, and be distinct from site-specific enrichments of hydrocarbons of biological origins.
Analysis
[edit]Substrates need to be prepared and analyzed in a specific way to elucidate site specific isotope enrichments. This requires clean separation of the compound of interest from the original sample, which can require a variety of different preparatory chemistries. Once isolated, position-specific isotope enrichments can be analyzed with a variety of instruments, which all have different advantages and provide varying degrees of precision.
Enzymatic Reaction
[edit]To measure the kinetic isotope effects of enzymatic reactions, biochemists perform in vitro experiments with enzymes and substrates. The goal of these experiments is to measure the difference in the enzymatic reaction rates for the monoisotopic substrate and the substrate with one rare isotope.[5] There are two popularly used techniques in these experiments: Internal competition studies and direct comparison experiments. Both measure position-specific isotope effects.
Direct Comparison
[edit]Direct comparison experiments are primarily used for measuring hydrogen/deuterium isotope effects in enzymatic reactions. The monoisotopic substrate and a deuterated form of the substrate are separately exposed to the enzyme of interest over a range of concentrations. The Michaelis-Menten kinetic parameters for both substrates are determined and the position-specific isotope effect at the site of deuteration is expressed as the ratio of the monoisotopic rate constant over the rare isotope rate constant.[7]
Internal Competition
[edit]For isotopes of elements like carbon and sulfur, the difference in kinetic parameters is too small, and the measurement precision too low, to measure an isotope effect by directly comparing the rates of the monoisotopic and rare isotope substrates. Instead, the two are mixed together using the natural abundance of stable isotopes in molecules. The enzyme is exposed to both isotopes simultaneously and its preference for the light isotope is analyzed by collecting the product of the reaction and measuring its isotope composition. For example, if an enzyme removes a carbon from a molecule by turning it into carbon dioxide, that carbon dioxide product can be collected and measured on an Isotope Ratio Mass Spectrometer for its carbon isotope composition. If the carbon dioxide has less 13C than the substrate mixture, the enzyme has preferentially reacted with the substrate that has a 12C at the site that is decarboxylated. In this way, internal competition experiments are also position-specific. If only the CO2 is measured, then only the isotope effect on the site of decarboxylation is recorded.[5]
Chemical degradation
[edit]Before the advent of technologies that analyze whole molecules for their intramolecular isotopic structure, molecules were sequentially degraded and converted to CO2 and measured on an Isotope Ratio Mass Spectrometer, revealing position-specific 13C enrichments.
Ninhydrin Reaction
[edit]In 1961, Abelson and Hoering developed a technique for removing the carboxylic acid of amino acids using the ninhydrin reaction. This reaction converts the carboxylic acid to a molecule of CO2 which is measured via an Isotope Ratio Mass Spectrometer.[1]
Ozonolysis Reaction
[edit]Lipids are of particular interest to stable isotope geochemists because they are preserved in rocks for millions of years. Monson & Hayes used ozonolysis to characterize the position-specific isotope abundances of unsaturated fatty acids, turning different carbon positions into carbon dioxide. Using this technique, they directly measured an isotopic pattern in fatty acids that had been predicted for years.[8]
Preparatory Chemistry
[edit]Derivatization
[edit]In some cases, additional functional groups will need to be added to molecules to facilitate the other separation and analysis methods. Derivatization can change the properties of an analyte; for instance, it would make a polar and non-volatile compound non-polar and more volatile, which would be necessary for analysis in certain types of chromatography. It is important to note, however, that derivatization is not ideal for site-specific analyses as it adds additional elements that must be accounted for in analyses.
Chromatography
[edit]Chromatography facilitates separation of distinct molecules within a mixture based on their respective chemical properties, and how those properties interact with the substrate coating the chromatographic column. This separation can happen “on-line,” during the measurement itself, or prior to measurements to isolate a pure compound. Gas and liquid chromatography have distinct advantages, based on the molecules of interest. For example, aqueously soluble molecules are more easily separated with liquid chromatography, while volatile, nonpolar molecules like propane or ethane are separated with gas chromatography.
Instrumental Analysis
[edit]A variety of different instruments can be used to perform position-specific isotope analysis, and each have distinct advantages and drawbacks. Many of them require comparison the sample of interest to a standard of known isotopic composition; fractionation within the instrument and variation of instrumental conditions over time can affect accuracy of individual measurements if not standardized.
GC-IRMS and LC-MS
[edit]Initial measurements of position specific isotope enrichments were measured using isotope ratio mass spectrometry in which sites on a molecule were first degraded to CO2, the CO2 was captured and purified, and then the CO2 was measured for its isotope composition on an Isotope Ratio Mass Spectrometer (IRMS). Py-GC-MS was also used in these experiments to degrade molecules even further and characterize their intramolecular isotopic distributions.[1] Both GC-MS and LC-MS are capable of characterizing position specific isotope enrichments in isotopically labelled molecules. In these molecules, 13C is so abundant that it can be seen on a mass spectrometer with low sensitivity. The resolution of these instruments can distinguish two molecules with a 1 Dalton difference in their molecular masses; however, this difference could arise from the addition of many rare isotopes (17O, 13C, 2H, etc.). For this reason, mass spectrometers using quadrupoles or time-of-flight detection techniques cannot be used for measuring position-specific enrichments at natural abundances.
Spectroscopy
[edit]Laser spectroscopy can be used to measure isotope enrichments of gases in the environment. Laser spectroscopy takes advantage of the different vibrational frequencies of isotopologues which cause them to absorb different wavelengths of light. Transmission of light through the gaseous sample at a controlled temperature can be quantitatively converted into a statement about isotopic composition. For N2O, these measurements can determine the position specific isotope enrichments of (15N.[9] These measurements are fast and can reach relatively good precision (1-10 per mille). It is used to characterize environmental gas fluxes, and effects on these fluxes.[10] This method is limited to measurement and characterization of gases.
Nuclear magnetic resonance (NMR)
[edit]Nuclear magnetic resonance observes small differences in molecular reactions to oscillating magnetic fields. It is able to characterize atoms with active nuclides that have a non-zero nuclear spin (e.g., 13C, 1H, 17O 35Cl, 15N, 37Cl), which makes it particularly useful for identifying certain isotopes. In typical proton or 13C NMR, the chemical shifts of protiums (1H) and carbon-13 atoms within a molecule are measured, respectively, as they are excited by a magnetic field and then relax with a diagnostic resonance frequency. With site specific natural isotope fractionation (SNIF) NMR, the relaxation resonances of the deuterium and 13C atoms.[11] NMR does not have the sensitivity to detect isotopologues with multiple rare isotopes. The only peaks that appear in a SNIF-NMR spectra are those of the isotopologues with a single rare isotope. Since the instrument is only measuring the resonances of the rare isotopes, each isotopologue will have one peak. For example, a molecule with six chemically unique carbon atoms will have six peaks in a 13C SNIF NMR spectrum. The site of 13C substitution can be determined by the chemical shift of each of the peaks. As a result, NMR is able to identify site specific isotope enrichments within molecules.[11][12]
Orbitrap Mass Spectrometry
[edit]
The Orbitrap is a high-resolution Fourier transform mass spectrometer that has recently been adapted to allow for site-specific analyses.[2] Molecules introduced into the Orbitrap are fragmented, accelerated, and analyzed. Because the Orbitrap characterizes molecular masses by measuring oscillations at radio frequencies, it is able to reach very high levels of precision, depending on measurement method (i.e., down to 0.1 per mille for long integration times). It is significantly faster than site-specific isotope measurements that can be performed using NMR, and can measure molecules with different rare isotopes but the same nominal mass at natural abundances (unlike GC and LCMS). It is also widely generalizable to molecules that can be introduced via gas or liquid solvent.[2] Resolution of the Orbitrap is such that nominal isobars (e.g., 2H versus 15N versus 13C enrichments) can be distinguished from one another, and so molecules do not need to be converted into a homogeneous substrate to facilitate isotope analysis. Like other isotope measurements, measurements of site-specific enrichments on the Orbitrap should be compared to a standard of known composition.[2]
Case studies
[edit]To illustrate the utility of position-specific isotope enrichments, several case studies are described below in which scientists used position-specific isotope analyses to answer important questions about biochemistry, pollution, and climate.
Phosphoenolpyruvate carboxylase
[edit]Phosphoenolpyruvate carboxylase (PEPC) is an enzyme that combines bicarbonate and phosphoenolpyruvate (PEP) to form the four-carbon acid, oxaloacetate. It is an important enzyme in C4 photosynthesis and anaplerotic pathways.[13] It is also responsible for the position-specific enrichment of oxaloacetate, due to the equilibrium isotope effect of converting the linear molecule CO2 into the trigonal planar molecule HCO3-, which partitions 13C into bicarbonate.[14] Inside the PEPC enzyme, H12CO3- reacts 1.0022 times faster than H13CO3- so that PEPC has a 0.22% kinetic isotope effect.[15] This is not enough to compensate for the 13C enrichment in bicarbonate. Thus, oxaloacetate is left with a 13C-enriched carbon at the C4 position. However, the C1 site experiences a small inverse secondary isotope effect due to its bonding environment in the transition state, leaving the C1 site of oxaloacetate enriched in 13C.[16] In this way, PEPC simultaneously partitions 12C into the C4 site and 13C into the C1 site of oxaloacetate, an example of multiple position-specific isotope effects.
Amino acids
[edit]The first paper on site-specific enrichment used the ninhydrin reaction to cleave the carboxyl site off alpha-amino acids in photosynthetic organisms.[1] The authors demonstrated an enriched carboxyl site relative to the bulk δ13C of the molecules, which they attribute to uptake of heavier CO2 through the Calvin cycle.[1] A recent study applied similar theory to understand enrichments in methionine, which they suggested would be powerful in origin and synthesis studies.[17]
Carbohydrates
[edit]
In 2012, a team of scientists used NMR spectroscopy to measure all of the position-specific carbon isotope abundances of glucose and other sugars. It was shown that the isotope abundances are heterogeneous. Different portions of the sugar molecules are used for biosynthesis based on the metabolic pathway an organism uses.[12] Therefore, any interpretations of position-specific isotopes of molecules downstream of glucose have to consider this intramolecular heterogeneity.
Glucose is the monomer of cellulose, the polymer that makes plants and trees rigid. After the advent of position-specific analyses of glucose, biogeochemists from Sweden looked the concentric tree rings of a Pinus nigra that recorded yearly growth between 1961 and 1995. They digested the cellulose down to its glucose units and used NMR spectroscopy to analyze its intramolecular isotopic patterns. They found correlations with position-specific isotope enrichments that were not apparent with whole molecule carbon isotope analysis of glucose. By measuring position-specific enrichments in the 6-carbon glucose molecule, they gathered six times more information from the same sample.[18]
Fatty acids
[edit]The biosynthesis of fatty acids begins with acetyl-CoA precursors that are brought together to make long straight chain lipids. Acetyl-CoA is produced in aerobic organisms by pyruvate dehydrogenase, an enzyme that has been shown to express a large, 2.3% isotope effect on the C2 site of pyruvate and a small fractionation on the C3 site.[19] These become the odd and even carbon positions of fatty acids respectively and in theory would result in a pattern of 13C depletions and enrichments at odd and even positions, respectively. In 1982, Monson and Hayes developed technology for measuring the position specific carbon isotope abundances of fatty acids. Their experiments on Escherichia coli revealed the predicted relative 13C enrichments at odd numbered carbon sites.[20] However, this pattern was not found in Saccharomyces cerevisiae that were fed glucose. Instead, its fatty acids were 13C enriched at the odd positions.[8] This has been interpreted as either a product of isotope effects during fatty acid degradation or the intramolecular isotopic heterogeneity of glucose that ultimately is reflected in the position-specific patterns of fatty acids.[21]
Nitrous Oxide
[edit]Site specific isotope enrichments of N2O is measured in the environment to help disentangle microbial sources and sinks in the environment. Different isotopologues of N2O absorb light at different wavelengths. Laser spectroscopy converts these differences as it scans across wavelengths to measure the abundance of 14N-15N-16O vs. 15N-14N-16O, a distinction that is impossible on other instruments. These measurements have achieved very high precision, down to 0.2 per mille.[10]
Environmental pollutants
[edit]Position-specific isotopes can be used to trace environmental pollutants through local and global environment.[22] This is specifically useful as heavy isotopes are often used to synthesize chemicals and then will get incorporated into the natural environment through biodegradation. Thus, tracing position-specific isotopes in the environment can help trace the movement of these pollutants and chemical products.
Case study conclusions
[edit]These case studies represent some potential applications for position specific isotope analysis, but certainly not all. The opportunities for samples to measure and processes to characterize are virtually unlimited, and new methodological developments will help make these measurements possible going forward.
References
[edit]- ^ a b c d e Abelson, Philip H. Hoering, T. C. CARBON ISOTOPE FRACTIONATION IN FORMATION OF AMINO ACIDS BY PHOTOSYNTHETIC ORGANISMS*. OCLC 678738249.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ^ a b c d Eiler, J. Cesar, J. Chimiak, L. Dallas, B. Grice, Kliti Griep-Raming, J. Juchelka, D. Kitchen, N. Lloyd, M. Makarov, A. Robins, R. Schwieters, J. (2017). Analysis of molecular isotopic structures at high precision and accuracy by Orbitrap mass spectrometry. Wiley InterScience. OCLC 1033992479.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ^ Hayes, John M. (2001-12-31), Valley, John W; Cole, David R (eds.), "3. Fractionation of Carbon and Hydrogen Isotopes in Biosynthetic Processes", Stable Isotope Geochemistry, De Gruyter, pp. 225–278, doi:10.1515/9781501508745-006, ISBN 978-1-5015-0874-5
- ^ a b Hermes, Jeffrey D.; Morrical, Scott W.; O'Leary, Marion H.; Cleland, W. W. (November 1984). "Variation of transition-state structure as a function of the nucleotide in reactions catalyzed by dehydrogenases. 2. Formate dehydrogenase". Biochemistry. 23 (23): 5479–5488. doi:10.1021/bi00318a016. ISSN 0006-2960. PMID 6391544.
- ^ a b c Wallace Cleland, W (November 2005), "Enzyme Mechanisms from Isotope Effects", Isotope Effects In Chemistry and Biology, CRC Press, pp. 915–930, doi:10.1201/9781420028027.ch37 (inactive 2024-11-11), ISBN 978-0-8247-2449-8
{{citation}}: CS1 maint: DOI inactive as of November 2024 (link) - ^ a b Suda, Konomi; Gilbert, Alexis; Yamada, Keita; Yoshida, Naohiro; Ueno, Yuichiro (June 2017). "Compound– and position–specific carbon isotopic signatures of abiogenic hydrocarbons from on–land serpentinite–hosted Hakuba Happo hot spring in Japan". Geochimica et Cosmochimica Acta. 206: 201–215. Bibcode:2017GeCoA.206..201S. doi:10.1016/j.gca.2017.03.008. ISSN 0016-7037.
- ^ Northrop, Dexter B. (1975-06-17). "Steady-state analysis of kinetic isotope effects in enzymic reactions". Biochemistry. 14 (12): 2644–2651. doi:10.1021/bi00683a013. ISSN 0006-2960. PMID 1148173.
- ^ a b Kd, Monson; Jm, Hayes (1982-05-25). "Biosynthetic Control of the Natural Abundance of Carbon 13 at Specific Positions Within Fatty Acids in Saccharomyces cerevisiae. Isotopic Fractionation in Lipid Synthesis as Evidence for Peroxisomal Regulation". The Journal of Biological Chemistry. 257 (10): 5568–75. doi:10.1016/S0021-9258(19)83814-0. PMID 7040368.
- ^ Waechter, Helen; Mohn, Joachim; Tuzson, Bela; Emmenegger, Lukas; Sigrist, Markus W. (2008-06-06). "Determination of N_2O isotopomers with quantum cascade laser based absorption spectroscopy". Optics Express. 16 (12): 9239–44. Bibcode:2008OExpr..16.9239W. doi:10.1364/oe.16.009239. ISSN 1094-4087. PMID 18545636.
- ^ a b Mohn, J.; Tuzson, B.; Manninen, A.; Yoshida, N.; Toyoda, S.; Brand, W. A.; Emmenegger, L. (2012-07-11). "Site selective real-time measurements of atmospheric N2O isotopomers by laser spectroscopy". Atmospheric Measurement Techniques. 5 (7): 1601–1609. doi:10.5194/amt-5-1601-2012. hdl:11858/00-001M-0000-000F-EE6C-C. ISSN 1867-8548.
- ^ a b Martin, Gérard J.; Akoka, Serge; Martin, Maryvonne L. (2006), Webb, Graham A. (ed.), "SNIF-NMR—Part 1: Principles", Modern Magnetic Resonance, Springer Netherlands, pp. 1651–1658, doi:10.1007/1-4020-3910-7_185, ISBN 978-1-4020-3910-2
- ^ a b Gilbert, A.; Robins, R. J.; Remaud, G. S.; Tcherkez, G. G. B. (2012-10-16). "Intramolecular 13C pattern in hexoses from autotrophic and heterotrophic C3 plant tissues". Proceedings of the National Academy of Sciences. 109 (44): 18204–18209. doi:10.1073/pnas.1211149109. ISSN 0027-8424. PMC 3497804. PMID 23074255.
- ^ Melzer, Eva; O'Leary, Marion H. (1987-05-01). "Anapleurotic CO2 Fixation by Phosphoenolpyruvate Carboxylase in C3 Plants". Plant Physiology. 84 (1): 58–60. doi:10.1104/pp.84.1.58. ISSN 0032-0889. PMC 1056527. PMID 16665405.
- ^ Mook, W.G.; Bommerson, J.C.; Staverman, W.H. (May 1974). "Carbon isotope fractionation between dissolved bicarbonate and gaseous carbon dioxide". Earth and Planetary Science Letters. 22 (2): 169–176. Bibcode:1974E&PSL..22..169M. doi:10.1016/0012-821x(74)90078-8. ISSN 0012-821X.
- ^ O'Leary, Marion H.; Rife, James E.; Slater, Jonathan D. (December 1981). "Kinetic and isotope effect studies of maize phosphoenolpyruvate carboxylase". Biochemistry. 20 (25): 7308–7314. doi:10.1021/bi00528a040. ISSN 0006-2960. PMID 7317383.
- ^ Gawlita, Ewa; Caldwell, William S.; O'Leary, Marion H.; Paneth, Piotr; Anderson, Vernon E. (February 1995). "Kinetic Isotope Effects on Substrate Association: Reactions of Phosphoenolpyruvate with Phosphoenolpyruvate Carboxylase and Pyruvate Kinase". Biochemistry. 34 (8): 2577–2583. doi:10.1021/bi00008a023. ISSN 0006-2960. PMID 7873538.
- ^ Neubauer, Cajetan; Sweredoski, Michael J.; Moradian, Annie; Newman, Dianne K.; Robins, Richard J.; Eiler, John M. (November 2018). "Scanning the isotopic structure of molecules by tandem mass spectrometry". International Journal of Mass Spectrometry. 434: 276–286. Bibcode:2018IJMSp.434..276N. doi:10.1016/j.ijms.2018.08.001. ISSN 1387-3806. S2CID 105288391.
- ^ a b Wieloch, Thomas; Ehlers, Ina; Yu, Jun; Frank, David; Grabner, Michael; Gessler, Arthur; Schleucher, Jürgen (2018-03-22). "Intramolecular 13C analysis of tree rings provides multiple plant ecophysiology signals covering decades". Scientific Reports. 8 (1): 5048. doi:10.1038/s41598-018-23422-2. ISSN 2045-2322. PMC 5864875. PMID 29567963.
- ^ Melzer, Eva. (1987). Carbon isotope effects on the pyruvate dehydrogenase reaction and their importance for relative carbon-13 depletion in lipids. American Society for Biochemistry and Molecular Biology. OCLC 793267425.
- ^ Monson, K.David; Hayes, J.M (February 1982). "Carbon isotopic fractionation in the biosynthesis of bacterial fatty acids. Ozonolysis of unsaturated fatty acids as a means of determining the intramolecular distribution of carbon isotopes". Geochimica et Cosmochimica Acta. 46 (2): 139–149. Bibcode:1982GeCoA..46..139M. doi:10.1016/0016-7037(82)90241-1. ISSN 0016-7037.
- ^ Schmidt, Hanns-Ludwig (2003-12-01). "Fundamentals and systematics of the non-statistical distributions of isotopes in natural compounds". Naturwissenschaften. 90 (12): 537–552. Bibcode:2003NW.....90..537S. doi:10.1007/s00114-003-0485-5. ISSN 0028-1042. PMID 14676950. S2CID 26485693.
- ^ Schmidt, Torsten C.; Zwank, Luc; Elsner, Martin; Berg, Michael; Meckenstock, Rainer U.; Haderlein, Stefan B. (2004-01-01). "Compound-specific stable isotope analysis of organic contaminants in natural environments: a critical review of the state of the art, prospects, and future challenges". Analytical and Bioanalytical Chemistry. 378 (2): 283–300. doi:10.1007/s00216-003-2350-y. ISSN 1618-2650. PMID 14647941. S2CID 36636525.