
Nitrile

In organic chemistry, a nitrile is any organic compound that has a −C≡N functional group. The name of the compound is composed of a base, which includes the carbon of the −C≡N, suffixed with "nitrile", so for example CH3CH2C≡N is called "propionitrile" (or propanenitrile).[1] The prefix cyano- is used interchangeably with the term nitrile in industrial literature. Nitriles are found in many useful compounds, including methyl cyanoacrylate, used in super glue, and nitrile rubber, a nitrile-containing polymer used in latex-free laboratory and medical gloves. Nitrile rubber is also widely used as automotive and other seals since it is resistant to fuels and oils. Organic compounds containing multiple nitrile groups are known as cyanocarbons.
Inorganic compounds containing the −C≡N group are not called nitriles, but cyanides instead.[2] Though both nitriles and cyanides can be derived from cyanide salts, most nitriles are not nearly as toxic.
Structure and basic properties
[edit]The N−C−C geometry is linear in nitriles, reflecting the sp hybridization of the triply bonded carbon. The C−N distance is short at 1.16 Å, consistent with a triple bond.[3] Nitriles are polar, as indicated by high dipole moments. As liquids, they have high relative permittivities, often in the 30s.
History
[edit]The first compound of the homolog row of nitriles, the nitrile of formic acid, hydrogen cyanide was first synthesized by C. W. Scheele in 1782.[4][5] In 1811 J. L. Gay-Lussac was able to prepare the very toxic and volatile pure acid.[6] Around 1832 benzonitrile, the nitrile of benzoic acid, was prepared by Friedrich Wöhler and Justus von Liebig, but due to minimal yield of the synthesis neither physical nor chemical properties were determined nor a structure suggested. In 1834 Théophile-Jules Pelouze synthesized propionitrile, suggesting it to be an ether of propionic alcohol and hydrocyanic acid.[7] The synthesis of benzonitrile by Hermann Fehling in 1844 by heating ammonium benzoate was the first method yielding enough of the substance for chemical research. Fehling determined the structure by comparing his results to the already known synthesis of hydrogen cyanide by heating ammonium formate. He coined the name "nitrile" for the newfound substance, which became the name for this group of compounds.[8]
Synthesis
[edit]Industrially, the main methods for producing nitriles are ammoxidation and hydrocyanation. Both routes are green in the sense that they do not generate stoichiometric amounts of salts.
Ammoxidation
[edit]In ammoxidation, a hydrocarbon is partially oxidized in the presence of ammonia. This conversion is practiced on a large scale for acrylonitrile:[9]

In the production of acrylonitrile, a side product is acetonitrile. On an industrial scale, several derivatives of benzonitrile, phthalonitrile, as well as Isobutyronitrile are prepared by ammoxidation. The process is catalysed by metal oxides and is assumed to proceed via the imine.
Hydrocyanation
[edit]Hydrocyanation is an industrial method for producing nitriles from hydrogen cyanide and alkenes. The process requires homogeneous catalysts. An example of hydrocyanation is the production of adiponitrile, a precursor to nylon-6,6 from 1,3-butadiene:
- CH2=CH−CH=CH2 + 2 HC≡N → NC(CH2)4C≡N
From organic halides and cyanide salts
[edit]Two salt metathesis reactions are popular for laboratory scale reactions. In the Kolbe nitrile synthesis, alkyl halides undergo nucleophilic aliphatic substitution with alkali metal cyanides. Aryl nitriles are prepared in the Rosenmund-von Braun synthesis.
In general, metal cyanides combine with alkyl halides to give a mixture of the nitrile and the isonitrile, although appropriate choice of counterion and temperature can minimize the latter. An alkyl sulfate obviates the problem entirely, particularly in nonaqueous conditions (the Pelouze synthesis).[5]
Cyanohydrins
[edit]
The cyanohydrins are a special class of nitriles. Classically they result from the addition of alkali metal cyanides to aldehydes in the cyanohydrin reaction. Because of the polarity of the organic carbonyl, this reaction requires no catalyst, unlike the hydrocyanation of alkenes. O-Silyl cyanohydrins are generated by the addition trimethylsilyl cyanide in the presence of a catalyst (silylcyanation). Cyanohydrins are also prepared by transcyanohydrin reactions starting, for example, with acetone cyanohydrin as a source of HCN.[10]
Dehydration of amides
[edit]Nitriles can be prepared by the dehydration of primary amides. Common reagents for this include phosphorus pentoxide (P2O5)[11] and thionyl chloride (SOCl2).[12] In a related dehydration, secondary amides give nitriles by the von Braun amide degradation. In this case, one C-N bond is cleaved.

Oxidation of amines
[edit]Numerous traditional methods exist for nitrile preparation by amine oxidation. [13] In addition, several selective methods have been developed in the last decades for electrochemical processes. [14]
From aldehydes and oximes
[edit]The conversion of aldehydes to nitriles via aldoximes is a popular laboratory route. Aldehydes react readily with hydroxylamine salts, sometimes at temperatures as low as ambient, to give aldoximes. These can be dehydrated to nitriles by simple heating,[15] although a wide range of reagents may assist with this, including triethylamine/sulfur dioxide, zeolites, or sulfuryl chloride. The related hydroxylamine-O-sulfonic acid reacts similarly.[16]

One-pot synthesis from aldehyde (Amberlyst is an acidic ion-exchange resin).

In specialised cases the Van Leusen reaction can be used. Biocatalysts such as aliphatic aldoxime dehydratase are also effective.
Sandmeyer reaction
[edit]Aromatic nitriles are often prepared in the laboratory from the aniline via diazonium compounds. This is the Sandmeyer reaction. It requires transition metal cyanides.[17]
- ArN+2 + CuC≡N → ArC≡N + N2 + Cu+
Other methods
[edit]- A commercial source for the cyanide group is diethylaluminum cyanide Et2AlCN which can be prepared from triethylaluminium and HCN.[18] It has been used in nucleophilic addition to ketones.[19] For an example of its use see: Kuwajima Taxol total synthesis
- Cyanide ions facilitate the coupling of dibromides. Reaction of α,α′-dibromoadipic acid with sodium cyanide in ethanol yields the cyano cyclobutane:[20]

- Aromatic nitriles can be prepared from base hydrolysis of trichloromethyl aryl ketimines (RC(CCl3)=NH) in the Houben-Fischer synthesis[21]
- Nitriles can be obtained from primary amines via oxidation. Common methods include the use of potassium persulfate,[22] Trichloroisocyanuric acid,[23] or anodic electrosynthesis.[24]
- α-Amino acids form nitriles and carbon dioxide via various means of oxidative decarboxylation.[25][26] Henry Drysdale Dakin discovered this oxidation in 1916.[27]
- From aryl carboxylic acids (Letts nitrile synthesis)

Reactions
[edit]Nitrile groups in organic compounds can undergo a variety of reactions depending on the reactants or conditions. A nitrile group can be hydrolyzed, reduced, or ejected from a molecule as a cyanide ion.
Hydrolysis
[edit]The hydrolysis of nitriles RCN proceeds in the distinct steps under acid or base treatment to first give carboxamides RC(O)NH2 and then carboxylic acids RC(O)OH. The hydrolysis of nitriles to carboxylic acids is efficient. In acid or base, the balanced equations are as follows:
- RC≡N + 2 H2O + HCl → RC(O)OH + NH4Cl
- RC≡N + H2O + NaOH → RC(O)ONa + NH3
Strictly speaking, these reactions are mediated (as opposed to catalyzed) by acid or base, since one equivalent of the acid or base is consumed to form the ammonium or carboxylate salt, respectively.
Kinetic studies show that the second-order rate constant for hydroxide-ion catalyzed hydrolysis of acetonitrile to acetamide is 1.6×10−6 M−1 s−1, which is slower than the hydrolysis of the amide to the carboxylate (7.4×10−5 M−1 s−1). Thus, the base hydrolysis route will afford the carboxylate (or the amide contaminated with the carboxylate). On the other hand, the acid catalyzed reactions requires a careful control of the temperature and of the ratio of reagents in order to avoid the formation of polymers, which is promoted by the exothermic character of the hydrolysis.[28] The classical procedure to convert a nitrile to the corresponding primary amide calls for adding the nitrile to cold concentrated sulfuric acid.[29] The further conversion to the carboxylic acid is disfavored by the low temperature and low concentration of water.
- RC≡N + H2O → RC(O)NH2
Two families of enzymes catalyze the hydrolysis of nitriles. Nitrilases hydrolyze nitriles to carboxylic acids:
- RC≡N + 2 H2O → RC(O)OH + NH3
Nitrile hydratases are metalloenzymes that hydrolyze nitriles to amides.
- RC≡N + H2O → RC(O)NH2
These enzymes are used commercially to produce acrylamide.
The "anhydrous hydration" of nitriles to amides has been demonstrated using an oxime as water source:[30]
- RC≡N + R'C(H)=NOH → RC(O)NH2 + R'C≡N
Reduction
[edit]Nitriles are susceptible to hydrogenation over diverse metal catalysts. The reaction can afford either the primary amine (RCH2NH2) or the tertiary amine ((RCH2)3N), depending on conditions.[31] In conventional organic reductions, nitrile is reduced by treatment with lithium aluminium hydride to the amine. Reduction to the imine followed by hydrolysis to the aldehyde takes place in the Stephen aldehyde synthesis, which uses stannous chloride in acid.
Deprotonation
[edit]Alkyl nitriles are sufficiently acidic to undergo deprotonation of the C-H bond adjacent to the C≡N group.[32][33] Strong bases are required, such as lithium diisopropylamide and butyl lithium. The product is referred to as a nitrile anion. These carbanions alkylate a wide variety of electrophiles. Key to the exceptional nucleophilicity is the small steric demand of the C≡N unit combined with its inductive stabilization. These features make nitriles ideal for creating new carbon-carbon bonds in sterically demanding environments.
Nucleophiles
[edit]The carbon center of a nitrile is electrophilic, hence it is susceptible to nucleophilic addition reactions:
- with an organozinc compound in the Blaise reaction
- with alcohols in the Pinner reaction.
- with amines, e.g. the reaction of the amine sarcosine with cyanamide yields creatine[34]
- with arenes to form ketones in the Houben–Hoesch reaction via an imine intermediate.
- with Grignard reagents to form primary ketimines in the Moureau-Mignonac ketimine synthesis.[35] While not a classical Grignard reaction, it may be considered one under broader modern definitions.
Miscellaneous methods and compounds
[edit]- In reductive decyanation the nitrile group is replaced by a proton.[36] Decyanations can be accomplished by dissolving metal reduction (e.g. HMPA and potassium metal in tert-butanol) or by fusion of a nitrile in KOH.[37] Similarly, α-aminonitriles can be decyanated with other reducing agents such as lithium aluminium hydride.[36]
- In the so-called Franchimont Reaction (developed by the Belgian doctoral student Antoine Paul Nicolas Franchimont (1844-1919) in 1872), an α-cyanocarboxylic acid heated in acid hydrolyzes and decarboxylates to a dimer.[38]
- Nitriles self-react in presence of base in the Thorpe reaction in a nucleophilic addition
- In organometallic chemistry nitriles are known to add to alkynes in carbocyanation:[39]

Complexation
[edit]Nitriles are precursors to transition metal nitrile complexes, which are reagents and catalysts. Examples include tetrakis(acetonitrile)copper(I) hexafluorophosphate ([Cu(MeCN)4]+) and bis(benzonitrile)palladium dichloride (PdCl2(PhCN)2).[40]
2.jpg)
Nitrile derivatives
[edit]Organic cyanamides
[edit]Cyanamides are N-cyano compounds with general structure R1R2N−C≡N and related to the parent cyanamide.[41]
Nitrile oxides
[edit]Nitrile oxides have the chemical formula RCNO. Their general structure is R−C≡N+−O−. The R stands for any group (typically organyl, e.g., acetonitrile oxide CH3−C≡N+−O−, hydrogen in the case of fulminic acid H−C≡N+−O−, or halogen (e.g., chlorine fulminate Cl−C≡N+−O−).[42]: 1187–1192
Nitrile oxides are quite different from nitriles: they are highly reactive 1,3-dipoles, and cannot be synthesized from the direct oxidation of nitriles.[43] Instead, they can be synthesised by dehydrogenation of oximes or by dehydration of nitroalkanes;[44]: 934–936 They are used in 1,3-dipolar cycloadditions,[42]: 1187–1192 such as to isoxazoles.[44]: 1201–1202 They undergo type 1 dyotropic rearrangement to isocyanates.[42]: 1700
The heavier nitrile sulfides are extremely reactive and rare, but temporarily form during the thermolysis of oxathiazolones. They react similarly to nitrile oxides.[45]
Occurrence and applications
[edit]Nitriles occur naturally in a diverse set of plant and animal sources. Over 120 naturally occurring nitriles have been isolated from terrestrial and marine sources. Nitriles are commonly encountered in fruit pits, especially almonds, and during cooking of Brassica crops (such as cabbage, Brussels sprouts, and cauliflower), which release nitriles through hydrolysis. Mandelonitrile, a cyanohydrin produced by ingesting almonds or some fruit pits, releases hydrogen cyanide and is responsible for the toxicity of cyanogenic glycosides.[46]
Over 30 nitrile-containing pharmaceuticals are currently marketed for a diverse variety of medicinal indications with more than 20 additional nitrile-containing leads in clinical development. The types of pharmaceuticals containing nitriles are diverse, from vildagliptin, an antidiabetic drug, to anastrozole, which is the gold standard in treating breast cancer. In many instances the nitrile mimics functionality present in substrates for enzymes, whereas in other cases the nitrile increases water solubility or decreases susceptibility to oxidative metabolism in the liver.[47] The nitrile functional group is found in several drugs.
-
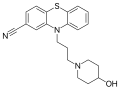
-
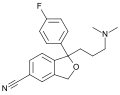 Structure of citalopram, an antidepressant drug of the selective serotonin reuptake inhibitor (SSRI) class.
Structure of citalopram, an antidepressant drug of the selective serotonin reuptake inhibitor (SSRI) class. -
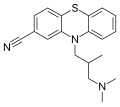 Structure of cyamemazine, an antipsychotic drug.
Structure of cyamemazine, an antipsychotic drug. -
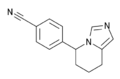 Structure of fadrozole, an aromatase inhibitor for the treatment of breast cancer.
Structure of fadrozole, an aromatase inhibitor for the treatment of breast cancer. -
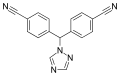 Structure of letrozole, an oral nonsteroidal aromatase inhibitor for the treatment of certain breast cancers.
Structure of letrozole, an oral nonsteroidal aromatase inhibitor for the treatment of certain breast cancers.





See also
[edit]- Protonated nitriles: Nitrilium
- Deprotonated nitriles: Nitrile anion
- Cyanocarbon
- Nitrile ylide
References
[edit]- ^ IUPAC Gold Book nitriles
- ^ NCBI-MeSH Nitriles
- ^ Karakida, Ken-ichi; Fukuyama, Tsutomu; Kuchitsu, Kozo (1974). "Molecular Structures of Hydrogen Cyanide and Acetonitrile as Studied by Gas Electron Diffraction". Bulletin of the Chemical Society of Japan. 47 (2): 299–304. doi:10.1246/bcsj.47.299.
- ^ See:
- Carl W. Scheele (1782) "Försök, beträffande det färgande ämnet uti Berlinerblå" (Experiment concerning the colored substance in Berlin blue), Kungliga Svenska Vetenskapsakademiens handlingar (Royal Swedish Academy of Science's Proceedings), 3: 264–275 (in Swedish).
- Reprinted in Latin as: "De materia tingente caerulei berolinensis" in: Carl Wilhelm Scheele with Ernst Benjamin Gottlieb Hebenstreit (ed.) and Gottfried Heinrich Schäfer (trans.), Opuscula Chemica et Physica (Leipzig ("Lipsiae"), (Germany): Johann Godfried Müller, 1789), vol. 2, pages 148–174.
- ^ a b David T. Mowry (1948). "The Preparation of Nitriles". Chemical Reviews. 42 (2): 189–283. doi:10.1021/cr60132a001. PMID 18914000.
- ^ Gay-Lussac produced pure, liquified hydrogen cyanide in: Gay-Lussac, J (1811). ""Note sur l'acide prussique" (Note on prussic acid)". Annales de chimie. 44: 128–133.
- ^ J. Pelouze (1834). "Notiz über einen neuen Cyanäther" [Note on a new cyano-ether]. Annalen der Pharmacie. 10 (3): 249. doi:10.1002/jlac.18340100302.
- ^ Hermann Fehling (1844). "Ueber die Zersetzung des benzoësauren Ammoniaks durch die Wärme (On the decomposition of ammonium benzoate by heat)". Annalen der Chemie und Pharmacie. 49 (1): 91–97. doi:10.1002/jlac.18440490106. On page 96, Fehling writes: "Da Laurent den von ihm entdeckten Körper schon Nitrobenzoyl genannt hat, auch schon ein Azobenzoyl existirt, so könnte man den aus benzoësaurem Ammoniak entstehenden Körper vielleicht Benzonitril nennen." (Since Laurent named the substance that was discovered by him "nitrobenzoyl" – also an "azobenzoyl" already exists – so one could name the substance that originates from ammonium benzoate perhaps "benzonitril".)
- ^ Pollak, Peter; Romeder, Gérard; Hagedorn, Ferdinand; Gelbke, Heinz-Peter (2000). "Nitriles". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a17_363. ISBN 3527306730.
- ^ Gregory, Robert J. H. (1999). "Cyanohydrins in Nature and the Laboratory: Biology, Preparations, and Synthetic Applications". Chemical Reviews. 99 (12): 3649–3682. doi:10.1021/cr9902906. PMID 11849033.
- ^ "ISOBUTYRONITRILE". Organic Syntheses. 25: 61. 1945. doi:10.15227/orgsyn.025.0061.
- ^ "2-ETHYLHEXANONITRILE". Organic Syntheses. 32: 65. 1952. doi:10.15227/orgsyn.032.0065.
- ^ Schümperli, Martin T.; Hammond, Ceri; Hermans, Ive (2021). "Developments in the Aerobic Oxidation of Amines". ACS Catal. 2 (6): 1108–1117. doi:10.1021/cs300212q.
- ^ Xu, Zhining; Kovács, Ervin (2024). "Beyond traditional synthesis: Electrochemical approaches to amine oxidation for nitriles and imines". ACS Org Inorg Au. 4 (5): 471–484. doi:10.1021/acsorginorgau.4c00025. PMC 11450732.
- ^ Chill, Samuel T.; Mebane, Robert C. (18 September 2009). "A Facile One-Pot Conversion of Aldehydes into Nitriles". Synthetic Communications. 39 (20): 3601–3606. doi:10.1080/00397910902788174. S2CID 97591561.
- ^ C. Fizet; J. Streith (1974). "Hydroxylamine-O-sulfonic acid: A convenient reagent for the oxidative conversion of aldehydes into nitriles". Tetrahedron Lett. (in German). 15 (36): 3187–3188. doi:10.1016/S0040-4039(01)91857-X.
- ^ "o-Tolunitrile and p-Tolunitrile" H. T. Clarke and R. R. Read Org. Synth. 1941, Coll. Vol. 1, 514.
- ^ W. Nagata and M. Yoshioka (1988). "Diethylaluminum cyanide". Organic Syntheses; Collected Volumes, vol. 6, p. 436.
- ^ W. Nagata, M. Yoshioka, and M. Murakami (1988). "Preparation of cyano compounds using alkylaluminum intermediates: 1-cyano-6-methoxy-3,4-dihydronaphthalene". Organic Syntheses
{{cite journal}}: CS1 maint: multiple names: authors list (link); Collected Volumes, vol. 6, p. 307. - ^ Reynold C. Fuson; Oscar R. Kreimeier & Gilbert L. Nimmo (1930). "Ring Closures in the Cyclobutane Series. II. Cyclization Of α,α′-Dibromo-Adipic Esters". J. Am. Chem. Soc. 52 (10): 4074–4076. doi:10.1021/ja01373a046.
- ^ J. Houben, Walter Fischer (1930) "Über eine neue Methode zur Darstellung cyclischer Nitrile durch katalytischen Abbau (I. Mitteil.)," Berichte der deutschen chemischen Gesellschaft (A and B Series) 63 (9): 2464 – 2472. doi:10.1002/cber.19300630920
- ^ Yamazaki, Shigekazu; Yamazaki, Yasuyuki (1990). "Nickel-catalyzed dehydrogenation of amines to nitriles". Bulletin of the Chemical Society of Japan. 63 (1): 301–303. doi:10.1246/bcsj.63.301.
- ^ Chen, Fen-Er; Kuang, Yun-Yan; Hui-Fang, Dai; Lu, Liang (2003). "A Selective and Mild Oxidation of Primary Amines to Nitriles with Trichloroisocyanuric Acid". Synthesis. 17 (17): 2629–2631. doi:10.1055/s-2003-42431.
- ^ Schäfer, H. J.; Feldhues, U. (1982). "Oxidation of Primary Aliphatic Amines to Nitriles at the Nickel Hydroxide Electrode". Synthesis. 1982 (2): 145–146. doi:10.1055/s-1982-29721. S2CID 97172564.
- ^ Hiegel, Gene; Lewis, Justin; Bae, Jason (2004). "Conversion of α-Amino Acids into Nitriles by Oxidative Decarboxylation with Trichloroisocyanuric Acid". Synthetic Communications. 34 (19): 3449–3453. doi:10.1081/SCC-200030958. S2CID 52208189.
- ^ Hampson, N; Lee, J; MacDonald, K (1972). "The oxidation of amino compounds at anodic silver". Electrochimica Acta. 17 (5): 921–955. doi:10.1016/0013-4686(72)90014-X.
- ^ Dakin, Henry Drysdale (1916). "The Oxidation of Amino-Acids to Cyanides". Biochemical Journal. 10 (2): 319–323. doi:10.1042/bj0100319. PMC 1258710. PMID 16742643.
- ^ Kukushkin, V. Yu.; Pombeiro, A. J. L. (2005). "Metal-mediated and metal-catalyzed hydrolysis of nitriles". Inorg. Chim. Acta. 358: 1–21. doi:10.1016/j.ica.2004.04.029.
- ^ Abbas, Khamis A. (1 January 2008). "Substituent Effects on the Hydrolysis of p-Substituted Benzonitriles in Sulfuric Acid Solutions at (25.0± 0.1) °C". Zeitschrift für Naturforschung A. 63 (9): 603–608. Bibcode:2008ZNatA..63..603A. doi:10.1515/zna-2008-0912. ISSN 1865-7109.
- ^ Dahye Kang; Jinwoo Lee; Hee-Yoon Lee (2012). "Anhydrous Hydration of Nitriles to Amides: p-Carbomethoxybenzamide". Organic Syntheses. 89: 66. doi:10.15227/orgsyn.089.0066.
- ^ Barrault, J.; Pouilloux, Y. (1997). "Catalytic Amination Reactions: Synthesis of fatty amines. Selectivity control in presence of multifunctional catalysts". Catalysis Today. 1997 (2): 137–153. doi:10.1016/S0920-5861(97)00006-0.
- ^ Arseniyadis, Siméon; Kyler, Keith S.; Watt, David S. (1984). "Addition and Substitution Reactions of Nitrile-Stabilized Carbanions". Organic Reactions. pp. 1–364. doi:10.1002/0471264180.or031.01. ISBN 978-0-471-26418-7.
- ^ Yang, Xun; Fleming, Fraser F. (2017). "C- and N-Metalated Nitriles: The Relationship between Structure and Selectivity". Accounts of Chemical Research. 50 (10): 2556–2568. doi:10.1021/acs.accounts.7b00329. PMID 28930437.
- ^ Smith, Andri L.; Tan, Paula (2006). "Creatine Synthesis: An Undergraduate Organic Chemistry Laboratory Experiment". J. Chem. Educ. 83 (11): 1654. Bibcode:2006JChEd..83.1654S. doi:10.1021/ed083p1654.
- ^ "Moureau-Mignonac Ketimine Synthesis". Comprehensive Organic Name Reactions and Reagents. Hoboken, NJ, USA: John Wiley & Sons, Inc. 15 September 2010. pp. 1988–1990. doi:10.1002/9780470638859.conrr446. ISBN 9780470638859.
- ^ a b The reductive decyanation reaction: chemical methods and synthetic applications Jean-Marc Mattalia, Caroline Marchi-Delapierre, Hassan Hazimeh, and Michel Chanon Arkivoc (AL-1755FR) pp. 90–118 2006 Article[permanent dead link]
- ^ Berkoff, Charles E.; Rivard, Donald E.; Kirkpatrick, David; Ives, Jeffrey L. (1980). "The Reductive Decyanation of Nitriles by Alkali Fusion". Synthetic Communications. 10 (12): 939–945. doi:10.1080/00397918008061855.
- ^ Franchimont, Antoine Paul Nicholas (1872). "Ueber die Dibenzyldicarbonsäure" [On 2,3-diphenylsuccinic acid]. Berichte der Deutschen Chemischen Gesellschaft. 5 (2): 1048–1050. doi:10.1002/cber.187200502138.
- ^ Yoshiaki Nakao; Akira Yada; Shiro Ebata & Tamejiro Hiyama (2007). "A Dramatic Effect of Lewis-Acid Catalysts on Nickel-Catalyzed Carbocyanation of Alkynes". J. Am. Chem. Soc. (Communication). 129 (9): 2428–2429. doi:10.1021/ja067364x. PMID 17295484.
- ^ Rach, S. F.; Kühn, F. E. (2009). "Nitrile Ligated Transition Metal Complexes with Weakly Coordinating Counteranions and Their Catalytic Applications". Chemical Reviews. 109 (5): 2061–2080. doi:10.1021/cr800270h. PMID 19326858.
- ^ March, Jerry (1992). Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (4th ed.). New York: Wiley. p. 436–7. ISBN 0-471-60180-2.
- ^ a b c Smith, Michael B.; March, Jerry (2007). March's Advanced Organic Chemistry (6th ed.). John Wiley & Sons. ISBN 978-0-471-72091-1.
- ^ Grundmann, Ch. (1970). "Nitrile oxides". In Rappoport, Zvi (ed.). The Chemistry of the Cyano Group. PATai's Chemistry of Functional Groups. p. 794. doi:10.1002/9780470771242.ch14. ISBN 978-0-471-70913-8.
- ^ a b Clayden, Jonathan; Greeves, Nick; Warren, Stuart; Wothers, Peter (2001). Organic Chemistry (1st ed.). Oxford University Press. ISBN 978-0-19-850346-0.
- ^ Argyropoulos, Nikolaos G. (1996). "1,4-Oxa/thia-2-azoles". In Katritzky, Alan R.; Rees, Charles W.; Scriven, Eric F. V. (eds.). Comprehensive Heterocyclic Chemistry. Vol. 4: Five-membered rings with more than two heteroatoms and fused carbocyclic derivatives. pp. 506–507. doi:10.1016/B978-008096518-5.00092-7. ISBN 978-0-08-096518-5.
- ^ Natural Product Reports Issue 5, 1999 Nitrile-containing natural products
- ^ Fleming, Fraser F.; Yao, Lihua; Ravikumar, P. C.; Funk, Lee; Shook, Brian C. (November 2010). "Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore". J Med Chem. 53 (22): 7902–17. doi:10.1021/jm100762r. PMC 2988972. PMID 20804202.
External links
[edit]- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "nitrile". doi:10.1351/goldbook.N04151
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "cyanide". doi:10.1351/goldbook.C01486