
Bismuthinidene

Bismuthinidenes are a class of organobismuth compounds, analogous to carbenes. These compounds have the general form R-Bi, with two lone pairs of electrons on the central bismuth(I) atom.[1] Due to the unusually low valency and oxidation state of +1, most bismuthinidenes are reactive and unstable,[2] though in recent decades, both transition metals and polydentate chelating Lewis base ligands have been employed to stabilize the low-valent bismuth(I) center through steric protection and π donation either in solution or in crystal structures.[1][3][4] Lewis base-stabilized bismuthinidenes adopt a singlet ground state with an inert lone pair of electrons in the 6s orbital.[1] A second lone pair in a 6p orbital and a single empty 6p orbital make Lewis base-stabilized bismuthinidenes ambiphilic.[3] Recently, a triplet bismuthinidene is reported by Cornella et al.[5]
Synthesis
[edit]Transition metal-stabilized bismuthinidene
[edit]The earliest examples of bismuthinidene complexes used transition metal chemistry to stabilize the Bi(I) center.[6][7][8][9] These methods generally leveraged the ability of simple bismuth(I) halides or methylbismuth to ligate to tungsten, manganese, and chromium carbonyl complexes. These complexes were occasionally found to oligomerize, forming Bi-Bi single or double bonds to form bismuthane or bismuthene moieties.[6][7][10] One of the first examples of a monomeric bismuthinidene was discovered by Balasz et al., who used R = 2-(dimethylaminomethyl)phenyl to chelate a Bi(I) center through a combination of strong C-Bi and weak N-Bi interactions.[11] Although the molecule quickly formed a cyclic oligomer, upon reaction with two equivalents of tungsten pentacarbonyl, monomeric crystalline RBi[W(CO)5]2 was isolated.[11]

N,C,N-coordinated bismuthinidene
[edit]Monomeric bismuthinidenes were not stabilized without the use of transition metal complexes until 2010, when Libor Dostál's research group reported the isolation of a bismuth(I) center stabilized only by the N,C,N-pincer ligand L = 2,6-bis[N-(2’,6’-dimethylphenyl)ketimino]phenyl.[3] This complex was first synthesized by reacting the precursor molecule LBiIIICl2 with two equivalents of the reducing agent K[B(iBu)3H] to yield isolable crystals of stable [C6H3-2,6-(C(Me)=N-2′,6′-Me2C6H3)2]Bi.[3] A slightly simpler N,C,N-coordinating ligand was soon used to create the bismuthinidene ArBi (Ar = C6H3-2,6-(CH=NtBu)2),[12][13] which became widely used in later bismuthinidene studies and is occasionally referred to as "Dostál's bismuthinidene".[14][15] In fact, many analogs of this compound have been synthesized, often replacing the imine tert-butyl groups with other bulky organic groups or replacing the two imine arms with disubstituted amine arms.[12][16]

N,C-coordinated bismuthinidene
[edit]Dostál's group later synthesized a monomeric bismuthinidene coordinated only by a bidentate N,C-coordinating ligand.[12] When the bismuth dichloride [C6H2-2-(CH=N-2’,6’-iPr2C6H3)-4,6-(tBu)2]BiCl2 is reduced by two equivalents of K[B(iBu)3H], isolable dark violet crystals of [C6H2-2-(CH=N-2’,6’-iPr2C6H3)-4,6-(tBu)2]Bi appear.[12] In contrast to the earlier transition metal-stabilized [2-(Me2NCH2)C6H4]Bi[W(CO)5]2, the tert-butyl group ortho to the bismuth atom in this N,C-coordinated bismuthinidene sterically block the partially empty p-type orbital on the bismuth atom, kinetically stabilizing it without the use of transition metals.[12] In addition, calculated nucleus-independent chemical shift indices (NICS) and anisotropy of current-induced density (ACID) analysis show that the BiC3N ring of the molecule was stabilized by a certain degree of aromatic character due and may be classified as a benzazabismole to the delocalization of six π electrons, despite the nominally dative Bi-N bond.[4][12] Unlike N,C,N-coordinated bismuthinidenes, this N,C-coordinated species requires the pendant nitrogen atom to be in an imine group, as replacement of the Dipp-substituted imine arm with a diethyl-substituted amine arm resulted in rapid dimerization to a dibismuthene species.[12]

Carbene-stabilized bismuthinidene
[edit]In 2019, Wang et al. isolated a novel carbene-stabilized bismuthinidene with an exocyclic bismuth(I) center.[2] Phenylbismuth dichloride, stabilized by a diethyl/diisopropylphenyl-substituted cyclic alkyl amino carbene (Et2CAAC), reacts with one equivalent of the beryllium(0) complex Be(Et2CAAC)2 in toluene to give stable, isolable red crystals of the carbene-stabilized bismuthinidene (Et2CAAC)Bi-Ph. Despite the exposed, exocyclic bismuth(I) center, the compound can exist without dimerization for up to two weeks in the solid state. Density functional theory (DFT) calculations showed that this is a result of partial double bond character between the carbene carbon and the bismuth(I) center, wherein the p-type lone pair of electrons on the bismuth atom interact with the partially-filled p orbitals on the carbene carbon.[2] However, the charge on the bismuth atom as determined by natural population analysis (NPA) was much lower than in bismuth(III)-carbon bonds, supporting the compound's classification as a bismuthinidene.[2]

Structural properties
[edit]As with other heavier carbene analogs, the structural and electronic properties of bismuthinidenes are in large part driven by the inert-pair effect, wherein the large energy gap between the bismuth atom's 6s and 6p orbitals disfavors the formation of sp hybrid orbitals.[17] In stark contrast to their lighter congeners phosphinidenes, whose smaller phosphorus 3s-3p energy gap favors a triplet ground state,[18] bismuthinidenes generally have a singlet ground state on account of the larger bismuth 6s-6p energy gap.[1]
The structural and electronic properties of bismuthinidenes in general are clearly exemplified by Dostál's N,C,N-stabilized bismuthinidene, which is the most commonly used bismuthinidene in the literature to date. Optimization of the tert-butyl imino version of this compound at the M06/cc-pVTZ level of theory reveals that, as in other nontrigonal pnictogen compounds, the central bismuth atom is coplanar with the N,C,N-coordinating ligand, adopting a T-shaped C2v coordination mode.[12][19][20][21] The Wiberg bond index (WBI) between the bismuth and carbon atoms is 1.09, while the Bi-C bond distance is 2.2156 Å, slightly shorter than the sum of these atoms’ single-bonded covalent radii (Σcov(Bi,C) = 2.26 Å).[12][22] On the other hand, the WBI of the Bi-N bonds is only 0.34, and the Bi-N bond distance is 2.500 Å, significantly longer than the sum of these atoms’ covalent radii (Σcov(Bi,N) = 2.22 Å). This agrees with calculations based on the quantum theory of atoms in molecules (QTAIM), which show that the electron density at the bond critical point between Bi and N is only 0.049, significantly lower than the electron density of 0.114 at the Bi-C bond critical point.[12][23] Natural bond orbital (NBO) calculations show that these weaker dative bonds arise from weak σ donation of the nitrogen atoms’ lone pairs into an empty 6p orbital on the central bismuth atom.[12][24] These two nN → p*Bi interactions stabilize the bismuthinidene by as much as 382 kJ/mol. Additionally, the amount of sigma donation from the pendant nitrogen atoms may be increased or decreased by replacing the tert-butyl groups on the pendant nitrogen atoms with aryl groups containing electron-donating groups or electron-withdrawing groups, respectively.[12][21] One lone pair resides in the bismuth atom's 6s orbital and generally remains inert, while the other resides in the 6p orbital oriented perpendicular to the plane of the central ring, which also comprises the highest occupied molecular orbital (HOMO).
-
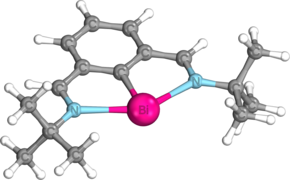 Optimized geometry of Dostál's bismuthinidene
Optimized geometry of Dostál's bismuthinidene -
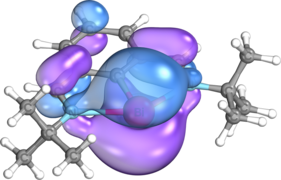 p-type lone pair on Bi
p-type lone pair on Bi -
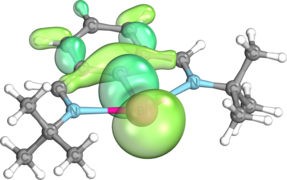 σ-bond between Bi and C
σ-bond between Bi and C -
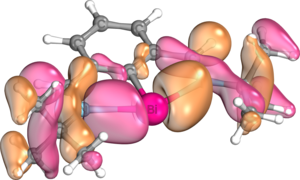 σ-donation of N lone pairs into empty p-type orbital on Bi
σ-donation of N lone pairs into empty p-type orbital on Bi -
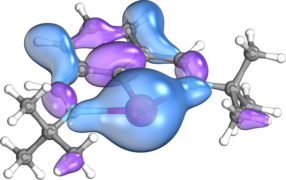 s-type lone pair on Bi
s-type lone pair on Bi





Reactivity
[edit]Theoretically, bismuthinidenes are both Lewis acidic and Lewis basic due to their empty and filled p-type orbitals, respectively. In practice, both N,C,N- and N,C-chelated bismuthinidenes lose much of their Lewis acidic character due to nN → p*Bi donor-acceptor interactions. However, the Lewis basicity of bismuthinidenes, particularly Dostál's N,C,N-coordinated bismuthinidene, allows them to cycle predictably between stable Bi(I) and Bi(III) oxidation states depending on the reaction conditions, allowing them to act as catalysts for a variety of different reactions, including transfer hydrogenations, deoxygenations, hydrodefluorinations, and dihydrogen reduction.[14][15][21][25][26] In addition, bismuthinidenes react intrinsically with certain alkyl halides, dichalcogenides, and alkynes to form Bi(III) species.[16][27][28][29]

Catalytic reactivity
[edit]The comparatively large covalent radius of bismuth results in weaker bonds between bismuth and other elements.[22][30] This is especially true of N,C,N-coordinated bismuthinidenes after oxidative addition of additional ligands to form trivalent bismuth(III) reactive intermediates, given the nN → p*Bi donation present in these species. This comparative weakness of the bismuth(III)-ligand bonds facilitates ligand exchange and ultimately reductive elimination to release the product, reform the bismuthinidene, and create a controllable Bi(I)/Bi(III) redox cycle that gives bismuthinidenes their own unique catalytic reactivity.[15][26]
Transfer hydrogenation of azoarenes
[edit]In 2019, Wang et al., who leveraged the catalytic activity of Dostál's bismuthinidene to catalyze a transfer hydrogenation reaction between ammonia-borane and azoarenes to form the corresponding arylhydrazines with good functional group tolerance.[26] The reaction's catalytic cycle proceeds through the oxidative addition of two hydrogen atoms from ammonia-borane to the bismuth(I) center, forming a highly unstable bismuthine intermediate.[26][31] Subsequent reductive elimination transfers the two hydrogen atoms across the pi bond of an azoarene molecule, restoring the bismuthinidene and forming arylhydrazine.[26][32] A similar bismuthinidene-catalyzed transfer hydrogenation reaction reduces nitroarenes to the corresponding aryl hydroxyl amines.[26]
Deoxygenation of nitrous oxide
[edit]
The Bi(I)/Bi(III) redox couple has also been applied to catalyze the deoxygenation of nitrous oxide.[14] When Dostál's bismuthinidene is exposed to gaseous N2O, the reaction mixture changes color from green to yellow and evolves dinitrogen gas. The color change is due to the formation of an arylbismuth oxide dimer with two μ-oxo bridging moieties forming a Bi2O2 center, consistent with the propensity of bismuth(III) oxides to spontaneously dimerize or polymerize.[10][14][33][34][35] However, a modified version of Dostál's bismuthinidene with ketimine arms and m-terphenyl substituents on the ketimine nitrogen atoms disfavors dimerization, instead forming a rare monomeric organobismuth(III) hydroxide upon reaction with N2O.[14] In either case, reduction of the product with pinacolborane (HBpin) returns the bismuth(III) centers to the bismuth(I) state and yields a mixture of HOBpin and (pinB)2O, completing the catalytic cycle.[14]
Hydrodefluorination of polyfluoroarenes
[edit]The electronic properties of the N,C,N-pincer ligand may be tuned with electron withdrawing groups to promote the reactivity of bismuthinidenes toward aryl C-F bonds.[15] One example is Phebox-Bi(I), an N,C,N-coordinated bismuthinidene stabilized by a 2,6-bis(oxazolinyl)phenyl (Phebox) pincer ligand. Unlike Dostál's bismuthinidene, which has only shown reactivity towards pentafluoropyridine, Phebox-Bi(I) has demonstrated a propensity to add to C-F bonds in a variety of perfluorinated arenes, including pentafluoropyridine, substituted pentafluorobenzenes, highly fluorinated phosphine compounds, octafluoronaphthalene, and decafluorobiphenyl.[15] After oxidative addition to the aryl C-F bond, the resulting Phebox-Bi(III)(fluoroaryl) fluoride intermediate may undergo ligand metathesis with diethylsilane, replacing the Bi(III)-F bond with a Bi(III)-H bond. The unstable Bi(III) hydride then undergoes aryl C-H reductive elimination, regenerating Phebox-Bi(I) and the hydrodefluorinated product.[15][31] The catalyst usually targets C-F bonds para to any electron-withdrawing substituents or heteroatoms on the fluorinated substrate. Reaction rates decrease significantly when the fluorinated substrate contains an electron-donating group.
Reduction of dihydrogen from acetic acid
[edit]Electrochemical studies indicate that Dostál's bismuthinidene may serve as an electrocatalyst for the formation of hydrogen gas.[25] Cyclic voltammetry and DFT calculations indicate that, under reducing conditions, acetic acid binds to the central bismuth atom, transferring a hydrogen atom to bismuth and generating an N,C,N-coordinated bismuth(III) acetate hydride intermediate.[25] Rearrangement of the acetate ligand from the equatorial to the axial position allows a second equivalent of acetic acid to bind to the bismuth center, eliminating H2 in the process.[25] Two-electron reduction releases acetate ligands, regenerating the bismuthinidene catalyst.
Intrinsic reactivity
[edit]Intrinsic bismuthinidene transformation reactions generally involve a double oxidation from the bismuth(I) redox state to the bismuth(III) redox state, generating unsymmetrically substituted trivalent bismuth(III) compounds that would have been difficult to synthesize through organometallic reagents.[27]
Oxidative addition toward alkyl halides and diphenyldichalcogenides
[edit]The low valency of bismuthinidenes renders them reactive toward carbon-polar group bonds.[27] Oxidative addition reactions between Dostál's bismuthinidene and primary C(sp3)-X bonds are particularly favorable for X = I or OTf, converting the bismuth(I) center to a bismuth(III) alkyl halide or alkyl triflate.[27] This is true even for longer fluorinated alkyl halides up to six carbon atoms in length. Steric hindrance prevents the activation of tert-butyl iodide by Dostál's bismuthinidene, although a metastable analog with amine pincer arms rather than imine pincer arms (Ar'Bi, where Ar' = 2,6-C6H3(CH2NMe2)2) does participate in oxidative addition even with bulky tertiary C-X bonds, likely because the increased rotational mobility of the amine arms allows them to rotate away from the incoming bulky alkyl group in the transition state.[27]
This metastable Ar'Bi, as well as N,C-stabilized bismuthinidene, are also reactive toward diphenyldichalcogenides.[16][28] While the former yields stable crystals of Ar'Bi(III)(EPh)2 (E = S, Se, Te) upon reaction with PhEEPh, the latter yields [2-C6H4(CH=NC6H3(i-Pr)2-2,6)]2Bi(III)(EPh) with two N,C ligands and only one phenyl chalcogenolate.[28] The N,C,N- and doubly N,C-coordinated bismuth(III) phenyl tellurolates are particularly unstable and decompose to form a mixture of products. Oxidative additions of N,C,N-coordinated bismuthinidene to diaryldisulfides are tolerant to a variety of aryl functional groups, including pyridyl, thiazolyl, thienyl, and aminophenyl groups.[16]

Hetero Diels-Alder reaction with alkynes
[edit]In 2019, Kořenková et al. discovered that Dostál's bismuthinidene behaves as a masked heterocyclic diene in the presence of the electron-deficient alkyne dimethyl acetylenedicarboxylate (DMAD), performing a hetero Diels-Alder [4+2] cycloaddition reaction to yield CO2Me-disubstituted 1-bisma-1,4-dihydro-iminonaphthalene, effectively converting one of the pendant imine arms of the bismuthinidene into a nitrogen-bridged bismacyclohexadiene, with the bismuth(III) atom serving as a bridgehead and the second imine arm largely losing coordination with the bismuth(III) center.[29] A similar cycloaddition reaction between Dostál's bismuthinidene and methyl propiolate yields an iminonaphthalene only as an intermediate, as the bridgehead bismuth atom is quickly attacked by a deprotonated second equivalent of methyl propiolate, breaking the Bi-N bond and yielding a boat-shaped bismacyclohexadiene moiety.[29] Though technically unbridged, the axial amine group on the bismacyclohexadiene ring remains datively coordinated to the bismuth heteroatom.
Transition metal chemistry
[edit]
Although N,C,N-coordinated bismuthinidenes are stable without transition metal coordination, they are reactive toward certain electron-deficient transition metal complexes and act as L-type donor ligands.[13] Upon addition of Dostál's bismuthinidene (ArBi; Ar = C6H3-2,6-(CH=NtBu)2) to solutions of dicobalt octacarbonyl or dimanganese decacarbonyl in toluene, isolable ionic crystals of [(ArBi)2Co(CO)3]+[Co(CO)4]- or [(ArBi)2Mn(CO)4]+[Mn(CO)5]- begin to form. These complexes show significant covalent interaction between the bismuth(I) atoms and the cobalt or manganese centers, though these bismuth-metal bonds are dative in character.[13] Because the bismuth-metal bonding consists almost entirely of σ-donation of the electrons in the p-type lone pair on bismuth into the dz2 orbital of the transition metal center, the ArBi units bond to the metal center in a side-on manner with C-Bi-Co or C-Bi-Mn bond angles close to 90°, such that the planes of the N,C,N ligands and those of the trigonal planar Co(CO)3 or square planar Mn(CO)4 moieties are all nearly parallel with each other. In this binding mode, the aromatic rings on the N,C,N ligands adopt a syn configuration stabilized by weak CH···CH and CH···O interactions.[13]
Dostál's bismuthinidene also binds to gold(I) centers stabilized by the N-heterocyclic carbene ligand IPr (1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene).[36] Ligand exchange with [Au(IPr)(ACN)]+[BF4]- yields the complex [Au(IPr)(ArBi)]+[BF4]- as a white powder. Though this complex is stabilized by the dative donation of electrons from Bi(I) into the empty 6s orbital of Au(I), this Bi(I) → Au(I) interaction is nevertheless stronger than any previously discovered Bi(III) → Au(I) interactions in which the bismuth atom acts as a donor. Furthermore, it represents the first stable, isolable complex containing Bi(I) → Au(I) interaction, which is thought to be enabled by the N,C,N-pincer ligand backbone.[36]
Similar reactivities have also been observed for the metastable version of Dostál's bismuthinidene containing amine pincer arms rather than imine pincer arms (Ar’Bi; Ar’ = C6H3-2,6-(CH2NMe2)2). Addition of this metastable bismuthinidene to THF solutions of M(CO)5 (where M = Cr, Mo, W) yields isolable crystals of [Ar’BiM(CO)5].[37] As before, the Ar’-Bi unit binds to the M(CO)5 moiety in a side-on fashion, with σ-donation from Bi(I) into the metal dz2 orbital. The reaction between Ar’-Bi and diiron nonacarbonyl likewise yields mostly [Ar’BiFe(CO)5], along with a small amount of [Ar’Bi(Fe(CO)4)2] as a minor product.[37] In fact, the reactivities of Dostál's bismuthinidene ArBi and its metastable analog Ar’-Bi toward transition metals are so similar that, upon reaction with Co2(CO)8, they form the analogous complexes [(ArBi)2Co(CO)3]+[Co(CO)4]- and [(Ar’Bi)2Co(CO)3]+[Co(CO)4]-, respectively, with similar binding modes, bond lengths, and bond angles.[37]
References
[edit]- ^ a b c d Mukhopadhyay, Deb Pratim; Schleier, Domenik; Wirsing, Sara; Ramler, Jacqueline; Kaiser, Dustin; Reusch, Engelbert; Hemberger, Patrick; Preitschopf, Tobias; Krummenacher, Ivo; Engels, Bernd; Fischer, Ingo; Lichtenberg, Crispin (2020). "Methylbismuth: an organometallic bismuthinidene biradical". Chemical Science. 11 (29): 7562–7568. doi:10.1039/D0SC02410D. PMC 7450715. PMID 32874526.
- ^ a b c d e Wang, Guocang; Freeman, Lucas A.; Dickie, Diane A.; Mokrai, Réka; Benkő, Zoltán; Gilliard, Robert J. (21 March 2019). "Isolation of Cyclic(Alkyl)(Amino) Carbene–Bismuthinidene Mediated by a Beryllium(0) Complex". Chemistry: A European Journal. 25 (17): 4335–4339. doi:10.1002/chem.201900458. PMC 6593863. PMID 30706565.
- ^ a b c d Šimon, Petr; de Proft, Frank; Jambor, Roman; Růžička, Aleš; Dostál, Libor (2 July 2010). "Monomeric Organoantimony(I) and Organobismuth(I) Compounds Stabilized by an NCN Chelating Ligand: Syntheses and Structures". Angewandte Chemie International Edition. 49 (32): 5468–5471. doi:10.1002/anie.201002209. PMID 20602393.
- ^ a b Dostál, Libor (December 2017). "Quest for stable or masked pnictinidenes: Emerging and exciting class of group 15 compounds". Coordination Chemistry Reviews. 353: 142–158. doi:10.1016/j.ccr.2017.10.009.
- ^ Pang, Yue; Nöthling, Nils; Leutzsch, Markus; Kang, Liqun; Bill, Eckhard; van Gastel, Maurice; Reijerse, Edward; Goddard, Richard; Wagner, Lucas; SantaLucia, Daniel; DeBeer, Serena; Neese, Frank; Cornella, Josep (2023-06-09). "Synthesis and isolation of a triplet bismuthinidene with a quenched magnetic response". Science. 380 (6649): 1043–1048. doi:10.1126/science.adg2833. ISSN 0036-8075.
- ^ a b Arif, Atta M.; Cowley, Alan H.; Norman, Nicholas C.; Pakulski, Marek (February 1985). "A tungsten-bismuth cluster featuring dibismuth as a four-electron donor and a bridging bismuthinidene". Journal of the American Chemical Society. 107 (4): 1062–1063. doi:10.1021/ja00290a054.
- ^ a b Arif, A. M.; Cowley, A. H.; Norman, N. C.; Pakulski, M. (December 1986). "Reactivity of bulky alkyldichlorostibines and alkyldichlorobismuthines toward [W(CO)5]2-: synthesis of compounds containing stibinidene, bismuthinidene, and dibismuth ligands". Inorganic Chemistry. 25 (27): 4836–4840. doi:10.1021/ic00247a012.
- ^ Davies, Simon J.; Compton, Neville A.; Huttner, Gottfried; Zsolnai, Laszlo; Garner, Stephanie E. (December 1991). "Synthesis and Reactivity of 'Bismuthinidene' Compounds and the Formation of Bi 1 Chelate Complexes". Chemische Berichte. 124 (12): 2731–2738. doi:10.1002/cber.19911241214.
- ^ Shieh, Minghuey; Cherng, Jiann-Jang; Lai, Yun-Wen; Ueng, Chuen-Her; Peng, Shie-Ming; Liu, Yi-Hung (4 October 2002). "Carbonylchromium derivatives of bismuth: new syntheses and relevance to cbond;o activation" (PDF). Chemistry. 8 (19): 4522–4527. doi:10.1002/1521-3765(20021004)8:19<4522::AID-CHEM4522>3.0.CO;2-7. PMID 12355541.
- ^ a b Tokitoh, Norihiro; Arai, Yoshimitsu; Okazaki, Renji; Nagase, Shigeru (4 July 1997). "Synthesis and Characterization of a Stable Dibismuthene: Evidence for a Bi-Bi Double Bond". Science. 277 (5322): 78–80. doi:10.1126/science.277.5322.78.
- ^ a b c Balazs, Lucia; Breunig, Hans J.; Lork, Enno; Silvestru, Cristian (April 2003). "Low-Valent Organobismuth Compounds with Intramolecular Coordination: cyclo -R 3 Bi 3 , cyclo -R 4 Bi 4 , RBi[W(CO) 5 ] 2 , and R 4 Bi 2 [R = 2-(Me 2 NCH 2 )C 6 H 4 ]". European Journal of Inorganic Chemistry. 2003 (7): 1361–1365. doi:10.1002/ejic.200390176.
- ^ a b c d e f g h i j k l m n Vránová, Iva; Alonso, Mercedes; Lo, Rabindranath; Sedlák, Robert; Jambor, Roman; Růžička, Aleš; Proft, Frank De; Hobza, Pavel; Dostál, Libor (16 November 2015). "From Dibismuthenes to Three- and Two-Coordinated Bismuthinidenes by Fine Ligand Tuning: Evidence for Aromatic BiC 3 N Rings through a Combined Experimental and Theoretical Study". Chemistry: A European Journal. 21 (47): 16917–16928. doi:10.1002/chem.201502724. PMID 26434943.
- ^ a b c d e Vránová, Iva; Alonso, Mercedes; Jambor, Roman; Růžička, Aleš; Erben, Milan; Dostál, Libor (23 May 2016). "Stibinidene and Bismuthinidene as Two-Electron Donors for Transition Metals (Co and Mn)". Chemistry: A European Journal. 22 (22): 7376–7380. doi:10.1002/chem.201601272. PMID 26994732.
- ^ a b c d e f Pang, Yue; Leutzsch, Markus; Nöthling, Nils; Cornella, Josep (18 November 2020). "Catalytic Activation of N 2 O at a Low-Valent Bismuth Redox Platform". Journal of the American Chemical Society. 142 (46): 19473–19479. doi:10.1021/jacs.0c10092. PMC 7677929. PMID 33146996.
- ^ a b c d e f g Pang, Yue; Leutzsch, Markus; Nöthling, Nils; Katzenburg, Felix; Cornella, Josep (18 August 2021). "Catalytic Hydrodefluorination via Oxidative Addition, Ligand Metathesis, and Reductive Elimination at Bi(I)/Bi(III) Centers". Journal of the American Chemical Society. 143 (32): 12487–12493. doi:10.1021/jacs.1c06735. PMC 8377712. PMID 34358426.
- ^ a b c d Šimon, Petr; Jambor, Roman; Růžička, Aleš; Dostál, Libor (September 2013). "Oxidative addition of organic disulfides to low valent N,C,N-chelated organobismuth(I) compound: Isolation, structure and coordination capability of substituted bismuth(III) bis(arylsulfides)". Journal of Organometallic Chemistry. 740: 98–103. doi:10.1016/j.jorganchem.2013.05.005.
- ^ Wheeler, Ralph A.; Kumar, P. N. V. Pavan (June 1992). "Stereochemically active or inactive lone pair electrons in some six-coordinate, group 15 halides". Journal of the American Chemical Society. 114 (12): 4776–4784. doi:10.1021/ja00038a049.
- ^ Lammertsma, Koop (2003). "Phosphinidenes". New Aspects in Phosphorus Chemistry III. Topics in Current Chemistry. Vol. 229. pp. 95–119. doi:10.1007/b11152. ISBN 978-3-540-00714-2.
- ^ Zhao, Yan; Truhlar, Donald G. (May 2008). "The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals". Theoretical Chemistry Accounts. 120 (1–3): 215–241. doi:10.1007/s00214-007-0310-x. S2CID 98119881.
- ^ Dunning, Thom H. (15 January 1989). "Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen". The Journal of Chemical Physics. 90 (2): 1007–1023. Bibcode:1989JChPh..90.1007D. doi:10.1063/1.456153.
- ^ a b c Gimferrer, Martí; Danés, Sergi; Andrada, Diego M.; Salvador, Pedro (6 December 2021). "Unveiling the Electronic Structure of the Bi(+1)/Bi(+3) Redox Couple on NCN and NNN Pincer Complexes". Inorganic Chemistry. 60 (23): 17657–17668. doi:10.1021/acs.inorgchem.1c02252. PMC 8653152. PMID 34766771.
- ^ a b Pyykkö, Pekka (19 March 2015). "Additive Covalent Radii for Single-, Double-, and Triple-Bonded Molecules and Tetrahedrally Bonded Crystals: A Summary". The Journal of Physical Chemistry A. 119 (11): 2326–2337. Bibcode:2015JPCA..119.2326P. doi:10.1021/jp5065819. PMID 25162610.
- ^ Bader, Richard F. W. (1 July 1991). "A quantum theory of molecular structure and its applications". Chemical Reviews. 91 (5): 893–928. doi:10.1021/cr00005a013.
- ^ Weinhold, Frank; Landis, Clark R. (2001). "Natural bond orbitals and extensions of localized bonding concepts". Chemistry Education Research and Practice. 2 (2): 91–104. doi:10.1039/B1RP90011K.
- ^ a b c d Xiao, Wang-Chuan; Tao, Yun-Wen; Luo, Geng-Geng (March 2020). "Hydrogen formation using a synthetic heavier main-group bismuth-based electrocatalyst". International Journal of Hydrogen Energy. 45 (15): 8177–8185. Bibcode:2020IJHE...45.8177X. doi:10.1016/j.ijhydene.2020.01.152. S2CID 213440010.
- ^ a b c d e f Wang, Feng; Planas, Oriol; Cornella, Josep (2019-03-13). "Bi(I)-Catalyzed Transfer-Hydrogenation with Ammonia-Borane". Journal of the American Chemical Society. 141 (10): 4235–4240. doi:10.1021/jacs.9b00594. PMC 6728098. PMID 30816708.
- ^ a b c d e Hejda, Martin; Jirásko, Robert; Růžička, Aleš; Jambor, Roman; Dostál, Libor (14 December 2020). "Probing the Limits of Oxidative Addition of C(sp 3 )–X Bonds toward Selected N,C,N -Chelated Bismuth(I) Compounds". Organometallics. 39 (23): 4320–4328. doi:10.1021/acs.organomet.0c00418. S2CID 225479032.
- ^ a b c Šimon, Petr; Jambor, Roman; Růžička, Aleš; Dostál, Libor (14 January 2013). "Oxidative Addition of Diphenyldichalcogenides PhEEPh (E = S, Se, Te) to Low-Valent CN- and NCN-Chelated Organoantimony and Organobismuth Compounds". Organometallics. 32 (1): 239–248. doi:10.1021/om3010383.
- ^ a b c Kořenková, Monika; Kremláček, Vít; Hejda, Martin; Turek, Jan; Khudaverdyan, Raffi; Erben, Milan; Jambor, Roman; Růžička, Aleš; Dostál, Libor (22 January 2020). "Hetero Diels–Alder Reactions of Masked Dienes Containing Heavy Group 15 Elements" (PDF). Chemistry: A European Journal. 26 (5): 1144–1154. doi:10.1002/chem.201904953. PMID 31769071. S2CID 208299504.
- ^ Lippert, E. (1960-08-21). "The Strengths of Chemical Bonds, von T. L. Cottrell. Butterworths Publications Ltd., London 1958. 2. Aufl., X, 317 S., geb.t—/32/—". Angewandte Chemie (in German). 72 (16): 602. Bibcode:1960AngCh..72..602L. doi:10.1002/ange.19600721618.
- ^ a b Balázs, Gábor; Breunig, Hans Joachim; Lork, Enno (1 June 2002). "Synthesis and Characterization of R 2 SbH, R 2 BiH, and R 2 Bi−BiR 2 [R = (Me 3 Si) 2 CH]". Organometallics. 21 (13): 2584–2586. doi:10.1021/om020202z.
- ^ Chu, Terry; Nikonov, Georgii I. (11 April 2018). "Oxidative Addition and Reductive Elimination at Main-Group Element Centers". Chemical Reviews. 118 (7): 3608–3680. doi:10.1021/acs.chemrev.7b00572. PMID 29558125.
- ^ Suzuki, Hitomi; Komatsu, Naoki; Ogawa, Takuji; Murafuji, Toshihiro; Ikegami, Tohru; Matano, Yoshihiro (2001-02-22). Organobismuth Chemistry. Elsevier. ISBN 978-0-08-053815-0.
- ^ Strîmb, Gabriela; Pöllnitz, Alpár; Raţ, Ciprian I.; Silvestru, Cristian (2015). "A general route to monoorganopnicogen( iii ) (M = Sb, Bi) compounds with a pincer (N,C,N) group and oxo ligands". Dalton Transactions. 44 (21): 9927–9942. doi:10.1039/C5DT00603A. PMID 25941006.
- ^ Matano, Yoshihiro; Nomura, Hazumi; Hisanaga, Teppei; Nakano, Haruyuki; Shiro, Motoo; Imahori, Hiroshi (1 November 2004). "Diverse Structures and Remarkable Oxidizing Ability of Triarylbismuthane Oxides. Comparative Study on the Structure and Reactivity of a Series of Triarylpnictogen Oxides". Organometallics. 23 (23): 5471–5480. doi:10.1021/om0494115.
- ^ a b Kořenková, Monika; Kremláček, Vít; Erben, Milan; Jirásko, Robert; De Proft, Frank; Turek, Jan; Jambor, Roman; Růžička, Aleš; Císařová, Ivana; Dostál, Libor (2018). "Heavier pnictinidene gold( i ) complexes". Dalton Transactions. 47 (41): 14503–14514. doi:10.1039/C8DT03022G. PMID 30283956.
- ^ a b c Vránová, Iva; Dušková, Tereza; Erben, Milan; Jambor, Roman; Růžička, Aleš; Dostál, Libor (May 2018). "Trapping of the N,C,N-chelated organobismuth(I) compound, [2,6-(Me2NCH2)2C6H3]Bi, by its coordination toward selected transition metal fragments". Journal of Organometallic Chemistry. 863: 15–20. doi:10.1016/j.jorganchem.2018.03.024.